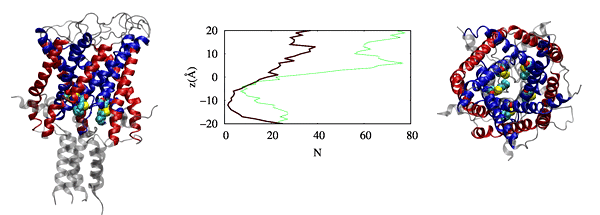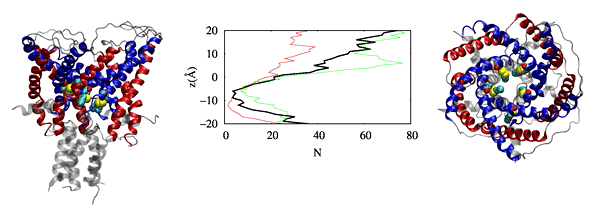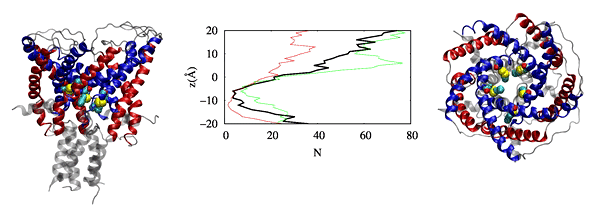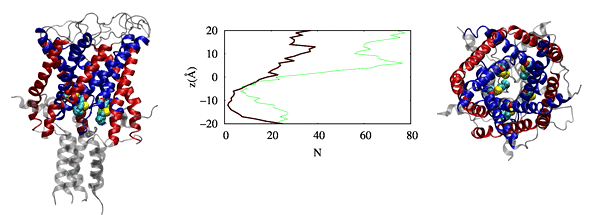
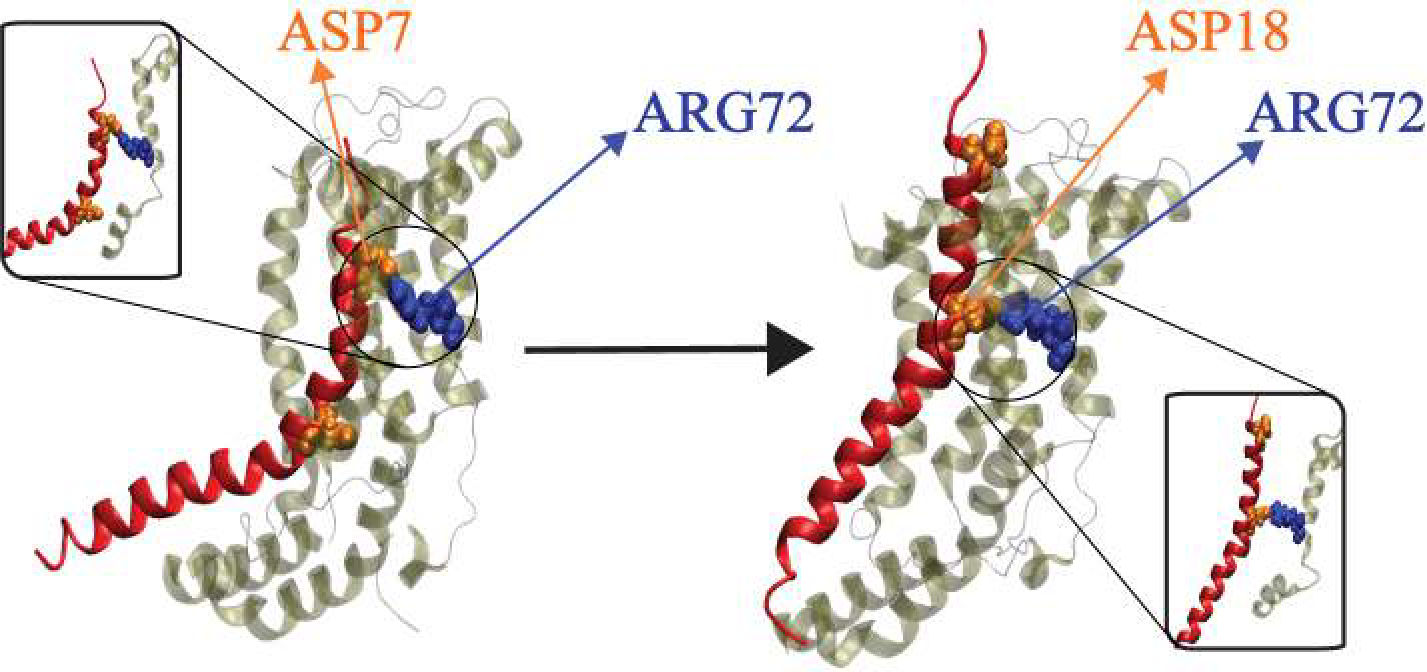
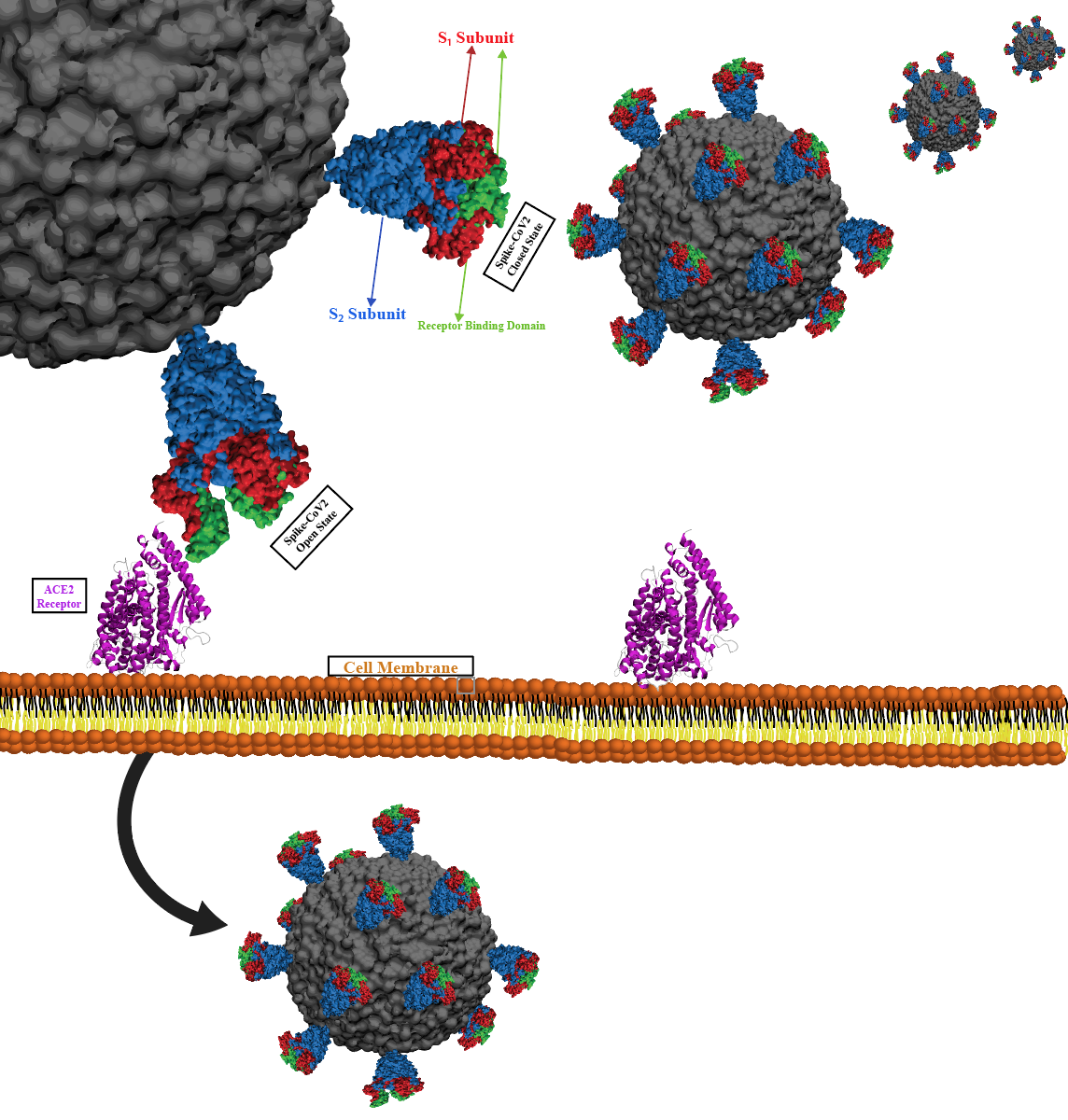
Physic-based computational estimation of binding affinities
This will allow researchers to estimate protein-ligand binding affinity

Examining experimentally the "millions" of available chemical compounds with drug-like properties is a formidable task. Researchers analyze drug databases computationally, filter them based on structure, chemical characteristics, and affinities towards target proteins, and then test the final, high-affinity molecules in the laboratory. Existing scoring systems find it difficult to account for entropy and desolvation effects, despite improvements in processing power. Even though computational approaches for incorporating protein flexibility in scoring are challenging and under development, they remain a potential option for protein-ligand binding affinity estimation. Quantitative agreement between computational and experimental estimates will result from the application of a binding affinity framework based on physics and employing unique free energy calculation methodologies. A method for estimating binding affinities that takes into account the conformational dynamics of both proteins and ligands and employs improved sampling methods within a unified framework could expedite the process of locating new drug-like molecules with accurate target binding affinities in enormous databases.
To calculate binding affinities, the proposed method employs cutting-edge technologies such as enhanced sampling generated from biased computer simulations,
a theoretical framework based on Riemannian geometry, and an orientation quaternion formalism in lieu of the conventional three Euler angles.
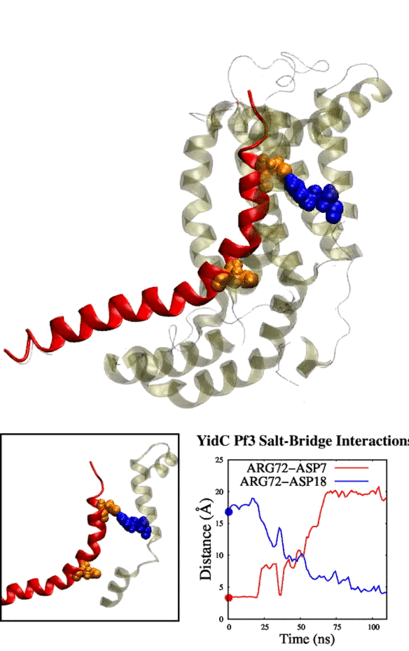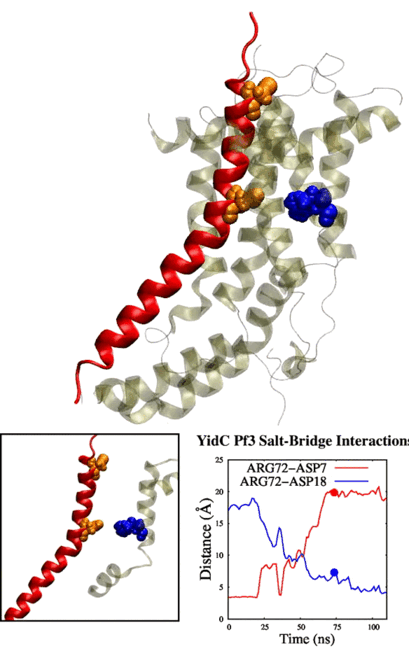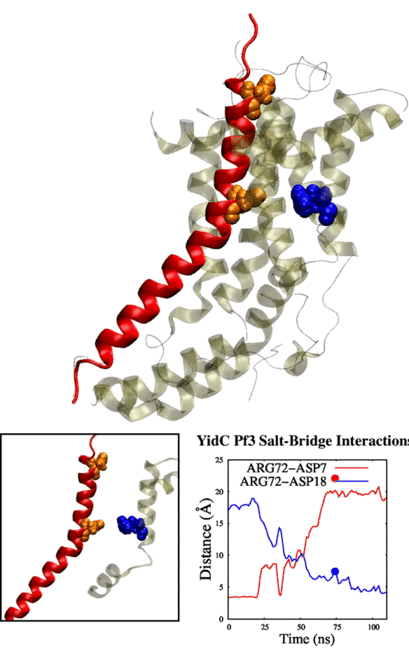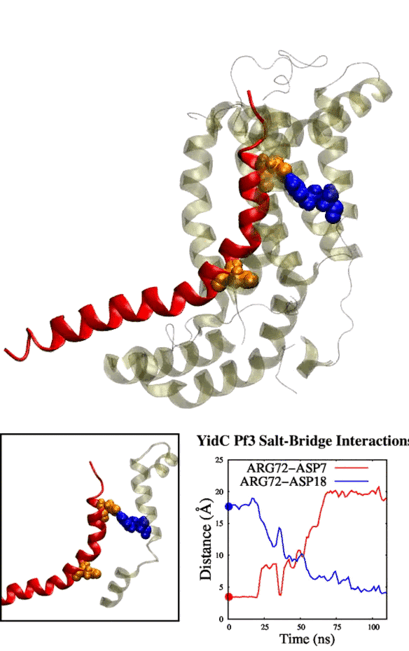
Our method generalizes and simplifies previously suggested stratification strategies that employ umbrella sampling or other enhanced sampling simulations with additional collective-variable-basedrestrictions. This method employs a versatile scheme that can be easily adapted to any system of interest. We estimate the binding affinity of human fibroblast growth factor 1 to heparin hexasaccharide using the crystal structure of the complex as the initial model and four variations of the proposed method to compare with the experimentally determined binding affinity obtained from isothermaltitration calorimetry experiments.
Research article:
Binding affinity estimation from restrained umbrella sampling simulations
Research article:
Binding affinity estimation from restrained umbrella sampling simulations